| Design | Participants | Care Providers | Outcome Assessors | Data Analysts |
|---|---|---|---|---|
| Open-label | Know | Know | Know | Know |
| Single-blind | Blinded | Know | Know | Know |
| Double-blind | Blinded | Blinded | Know | Know |
| Triple-blind | Blinded | Blinded | Blinded | Know |
| Analyst-blind | May or may not know | May or may not know | May or may not know | Blinded |
10 Randomized Controlled Trials
Let me tell you about a choice that saved millions of lives—not through a medical breakthrough that took decades to discover, but through a simple decision to test an idea rigorously.
In the 1970s, diarrheal disease was killing an estimated 4–5 million children each year, primarily from dehydration (Snyder et al., 1982). The standard treatment for severe dehydration was intravenous fluids—effective, but dependent on hospitals, trained staff, sterile equipment, and infrastructure that simply didn’t exist in most places where children were dying. When a child in a rural Bangladeshi village developed severe diarrhea, the nearest hospital might be hours away by foot. By the time families could reach help, it was often too late.
The physiological principles for an alternative had been taking shape since the 1960s: researchers had discovered that glucose and sodium are co-transported across the intestinal wall, meaning that a simple solution of salt, sugar, and clean water could be absorbed even during active diarrhea. But could something a mother mixed at home actually replace hospital IV therapy? Many clinicians were skeptical.
A 1978 Lancet editorial called the discovery of glucose-sodium cotransport “potentially the most important medical advance this century” (1978).
Researchers in South Asia, particularly at the Cholera Research Laboratory in Dhaka (now ICDDR,B), set out to answer that question with randomized controlled trials. They randomly assigned children with acute diarrhea to receive either oral rehydration therapy (ORT) or standard intravenous treatment and measured outcomes meticulously. The results were stunning: for mild to moderate dehydration, ORT worked remarkably well, dramatically reducing the need for IV therapy and preventing death from dehydration in the vast majority of cases. Severe dehydration still required IV fluids initially, but ORT could sustain children afterward—and, crucially, it could be delivered at home, in refugee camps, in villages with no hospital at all.
Dilip Mahalanabis’ use of ORT in refugee camps during the 1971 Bangladesh Liberation War demonstrated its life-saving potential in crisis conditions—even before the randomized trials were complete (Mahalanabis et al., 1973).
That evidence changed everything. Today, oral rehydration therapy is credited with saving more than 50 million lives since its widespread adoption (Fontaine et al., 2007; Nalin et al., 2018). Think about that number for a moment. Fifty million people—mostly children—who are alive because researchers had the courage and methodological rigor to test a simple idea through randomized controlled trials.
This is the power—and the purpose—of randomized controlled trials in global health. They allow us to distinguish between what we think might work and what actually does work when implemented in real-world conditions. They help us avoid wasting scarce resources on interventions that sound good but don’t deliver results. And sometimes, as with ORT, they reveal that the most effective solution isn’t the most technologically sophisticated one.
In this chapter, we’re going to explore how randomized controlled trials work, why they matter in global health settings, and how you can design and conduct them ethically and effectively. You’ll come to understand why randomization matters for establishing causality—and you’ll learn to design RCTs that address meaningful research questions, from selecting participants to determining appropriate control conditions. We’ll examine landmark trials that changed policy, navigate the ethical complexities of conducting research in low-resource settings, and develop practical skills for critically evaluating published studies and translating findings into policy recommendations.
10.1 Understanding Randomized Controlled Trials
WHAT IS RANDOMIZATION, AND WHY DOES IT MATTER?
You’ve encountered the term “randomized controlled trial”, or RCT, dozens of times so far in this book. You know RCTs are important. You know they’re considered the “gold standard” for evaluating interventions. But do you understand why randomization is so crucial?
A randomized controlled trial is a study design in which participants (or units like classrooms or villages) are randomly assigned to different groups—typically an intervention group that receives the treatment being tested and a control group that receives either a placebo, standard care, or no intervention. The key word here is “randomly.” Not based on which patients arrive first, not based on doctor preference, not based on disease severity, but based purely on chance, like flipping a coin.
Randomization is the process of assigning participants to study groups using chance alone, ensuring that each participant has a known probability (usually equal) of being assigned to any given group. This process is what distinguishes RCTs from other comparative study designs.
Why does randomization matter so much? Because it solves a problem that bedeviled medical research for centuries: how do we know whether an observed improvement in patients is due to the treatment we gave them, or due to something else entirely?
Consider this scenario. You’re evaluating a new community health worker program designed to improve child nutrition in rural Ethiopia. You implement the program in ten villages and compare child growth outcomes to ten other villages that didn’t receive the program. Six months later, you find that children in program villages have better nutrition indicators. Success, right?
Not so fast. What if the villages you selected for the program were closer to health facilities to begin with? What if they had better access to markets? What if community leaders in those villages were particularly motivated and would have implemented nutrition improvements even without your program? Any of these factors—what we call confounding variables—could explain the better nutrition outcomes you observed, rather than the program itself.
This is where randomization works its magic. When you randomly assign villages to receive the program or not, you ensure that all those potential confounding variables—distance to health facilities, market access, community leadership quality, baseline nutrition knowledge, wealth, education, and countless other factors you didn’t even think to measure—are distributed evenly across program and control villages on average. Some program villages might be close to health facilities and some far away, but the same will be true in control villages. The groups become comparable not through careful matching (which can only account for variables you know to measure) but through the beautiful mathematics of randomization, which accounts for everything—measured and unmeasured alike.
A BRIEF HISTORY: FROM SCURVY TO HIV PREVENTION
The history of randomized controlled trials in medicine is fascinating, though we’ll keep this brief because you’re here to learn how to do RCTs in global health, not to become a medical historian. But understanding where RCTs came from helps appreciate why they matter.
The roots of controlled experimentation in medicine are often traced to James Lind, a British naval surgeon who in 1747 tested six potential remedies for scurvy aboard HMS Salisbury. Lind selected twelve sailors with similar symptoms and deliberately assigned pairs to receive cider, sulfuric acid, vinegar, sea water, a medicinal paste, or citrus fruits. The sailors who received oranges and lemons recovered quickly; the others did not. It was a controlled comparison—Lind tried to match patients on severity—but it was not randomized. He chose who got what.
The first truly randomized clinical trial came two centuries later: the British Medical Research Council’s 1948 streptomycin trial for tuberculosis, designed by Austin Bradford Hill (Medical Research Council Streptomycin in Tuberculosis Trials Committee, 1948). That trial established the template for modern RCTs: centrally generated random allocation using sequentially numbered sealed envelopes—opened only after a patient was deemed eligible, to prevent foreknowledge of assignment—concurrent control groups, and systematic outcome assessment.
In global health, RCTs emerged somewhat later, initially focused on infectious disease prevention and treatment in low- and middle-income countries. The oral rehydration therapy trials I mentioned at the start of this chapter were pivotal—not just for their public health impact, but for demonstrating that rigorous RCTs could be conducted in resource-limited settings and generate evidence with immediate policy relevance.
More recently, RCTs in global health have expanded far beyond pharmaceutical interventions to test health system innovations, behavioral interventions, community health worker programs, mobile health technologies, and complex implementation strategies. The HIV Prevention Trials Network (HPTN) 052 study, conducted across nine countries in Asia, Africa, and the Americas, demonstrated that early antiretroviral treatment reduced HIV transmission in serodiscordant couples by 96% among phylogenetically linked infections—and by 89% when including all transmissions (Cohen et al., 2012). The trial provided pivotal evidence for treatment-as-prevention approaches, a paradigm shift made possible by rigorous multi-country RCT evidence.
Historical note: The HPTN 052 trial was stopped early by the Data Safety Monitoring Board because the benefit of early treatment was so clear that continuing the trial would have been unethical. This demonstrates how carefully monitored RCTs protect participant safety while generating definitive evidence.
THE CAUSAL INFERENCE SUPERPOWER OF RCTS
What makes RCTs special in the hierarchy of evidence is that they’re our best tool for establishing causality. Not just correlation, not just association, but actual cause-and-effect relationships.
Remember from Chapter 7 that establishing causation requires answering a counterfactual question: what would have happened to a child who received oral rehydration therapy if they had instead received IV fluids? We can’t know, because the fact is they received ORT—we only observe what actually happened. This missing data problem is the fundamental problem of causal inference: we never get to see both potential outcomes for the same person.
RCTs solve this problem—or more accurately, they solve it as well as is practically possible in the real world. By randomly assigning people to intervention and control groups, we create two groups that, on average, are comparable at baseline—not because we matched them on every characteristic, but because randomization ensures that any differences between groups are due to chance rather than systematic bias. Any differences in outcomes between these groups after the intervention can then be attributed to the intervention itself, because randomization ensured that the groups didn’t differ systematically in any other way.
Put another way, randomization lets us get as close as we’ll ever get to observing the same person both receiving and not receiving an intervention.
The Fundamental Problem of Causal Inference: For any individual, we can observe either what happens when they receive an intervention OR what happens when they don’t receive it, but never both. RCTs address this by ensuring that the group that receives the intervention is statistically equivalent to the group that doesn’t, so we can use the control group as a valid counterfactual for the intervention group.
This doesn’t mean RCTs are perfect. They’re not. Random assignment ensures balance on average across many trials or in large samples, but any particular trial might, by chance, have groups that differ in important ways. This is why we preregister our analyses and report all outcomes, even when results aren’t what we hoped.
RCTs remain our strongest tool for establishing that an intervention causes the outcomes we care about, rather than just being associated with them. Let’s learn how to design one.
10.2 Designing an RCT
Let’s turn to the practical question: how do you actually design one? This section will walk you through the key design decisions you’ll face, using real global health examples to illustrate the principles.
FORMULATING RESEARCH QUESTIONS WORTH ANSWERING
Every RCT begins with a research question. But not just any question—a question that’s both answerable through randomization and worth the substantial resources an RCT requires. Let’s use the PICO framework, introduced in Chapter 3, to structure the question itself—because for RCTs, clarity about your Population, Intervention, Comparison, and Outcome is what separates a vague idea from a testable hypothesis.
PICO stands for Population, Intervention, Comparison, Outcome. See Chapter 3 for a full walkthrough of both PICO and FINER.
- Population: Who are you studying? Be specific about age, setting, and health status.
- Intervention: What treatment, program, or exposure are you testing?
- Comparison: What are you comparing it to—usual care, placebo, an alternative intervention, nothing?
- Outcome: What will you measure to determine whether the intervention worked?
Let’s apply this to a real example. Suppose you’re interested in improving HPV vaccination rates among young women in rural India. The research literature shows low awareness of HPV and cervical cancer, high vaccine costs, and persistent safety concerns (Coursey et al., 2024). Some of those concerns trace back to a 2009–2010 incident in which seven girls died during a PATH HPV vaccination demonstration project; a government inquiry later determined that the deaths were unrelated to the vaccine, but the misconception persisted (Government of India, Ministry of Health and Family Welfare, 2011).
You could frame several different PICO questions:
| Question 1 | Question 2 | Question 3 | |
|---|---|---|---|
| Population | Young women aged 15–25 in rural Rajasthan | Same | Same |
| Intervention | Free HPV vaccination at community health centers | CHW-led education on vaccine safety | Varied vaccine attributes (cost, doses, side effects) in hypothetical scenarios |
| Comparison | Standard pricing | Standard information pamphlet | Across attribute levels |
| Outcome | Vaccination uptake at 6 months | Willingness to vaccinate (and actual uptake) | Stated preferences for vaccination |
All three are interesting and relevant. But notice how PICO forces you to be specific. Question 1 has a clean comparison and a measurable outcome—it’s ready for an RCT. Question 2 is promising but the intervention needs more definition: what exactly does the education session cover, and how long is it? Question 3 might be best answered through discrete choice experiments (a method we’ll discuss in a later chapter) rather than an RCT.
There’s no single “right” answer here—the best research question depends on your resources, timeline, policy context, and what evidence gaps are most critical to fill. But structuring your question with PICO forces the specificity that turns a good idea into a feasible trial.
Pause and reflect: Think about a global health intervention you’re interested in evaluating. Write down a potential RCT research question using PICO. Can you clearly state each element? If you’re struggling with the comparison or outcome, that’s a sign your question needs refining before you can design a trial around it.
RECRUITMENT AND ELIGIBILITY
Once you have a research question, you need to figure out who will be in your study and how you’ll get them there. This involves three decisions: defining eligibility criteria (who qualifies), planning recruitment (how you’ll find and enroll them), and thinking carefully about what your study sample means for the conclusions you can draw.
A point that’s easy to overlook: RCTs almost never involve random selection from a population. Random assignment determines which group participants are assigned to—treatment or control—but the people who end up in your trial are rarely a random sample of anyone. They’re volunteers who met your eligibility criteria, showed up at a participating site, and consented to participate. This means your trial has strong internal validity (randomization protects the comparison between groups) but the question of whether your results apply beyond your specific sample—transportability—is always open. We cover this in depth in Chapter 8.
Randomized assignment ≠ random selection. Randomization controls who gets the intervention. It does not control who enters the trial in the first place. This distinction matters enormously for interpreting results.
Eligibility criteria define the population for which the treatment effect is directly estimated. They are not just a logistical detail; they determine the trial’s target population.
Consider the Ethiopian oral cholera vaccine trial in Addis Ababa (Desai et al., 2015). The investigators enrolled healthy adults and children, excluding individuals with serious prior vaccine reactions, immunocompromising conditions, or recent febrile illness. These restrictions improved protocol safety and reduced biological heterogeneity, allowing for a cleaner assessment of immunogenicity.
But the effect estimate therefore applies most directly to relatively healthy individuals. The trial cannot, on its own, tell us how the vaccine performs in HIV-positive individuals, in people with malnutrition, or in populations with high comorbidity burdens—precisely the settings where cholera vaccination is often most needed.
This is not a simple tradeoff between internal and external validity. Restrictive criteria can reduce variability and simplify interpretation, but they also narrow the population for whom the causal effect is identified. Broader inclusion increases heterogeneity, yet it improves the ability to examine effect modification and to assess transportability to real-world populations.
In global health research, I generally advocate for inclusive eligibility criteria when ethically and operationally feasible. If comorbidities are common in the target population, excluding them at the design stage forces policymakers to extrapolate beyond the evidence. A trial can be statistically rigorous and still leave major uncertainty about who benefits—if it defines the eligible population too narrowly.
Recruitment is where many trials struggle. Enrollment projections are almost always optimistic. You need a clear plan: Where will you recruit? Through clinics, community outreach, health worker referrals? How will you ensure you’re reaching the population your research question is about, not just the population that’s easy to find? Community engagement and trusted local partnerships are often more effective than flyers and advertisements—and they yield samples that better reflect the people your intervention is intended to serve.
Plan to collect rich baseline data on participants so you can later examine heterogeneity in treatment effects (see the section on heterogeneity later in this chapter). If your intervention works differently for men and women, or for urban and rural participants, you want to know that—and you can only know it if your sample includes those subgroups and you measured the relevant characteristics.
SAMPLE SIZE: HOW MANY PARTICIPANTS DO YOU ACTUALLY NEED?
I devote a full chapter to sample size and statistical power later in this book, so here I’ll focus on the concepts that matter most at the design stage of an RCT.
The core idea is simple: your trial needs to be large enough to detect a real intervention effect if one exists, but not so large that you waste resources. Too small, and you’ll miss real effects (Type II error). Too large, and you’re spending money and participants’ time to achieve precision you don’t need—resources that could have funded other research or direct health services.
The most important parameter in any sample size calculation is the minimum detectable effect size (MDES)—the smallest effect your trial is powered to find. This is where you need to think carefully about what difference would be clinically or practically meaningful. If a new treatment reduces systolic blood pressure from 150 to 149 mmHg on average, is that worth the cost of implementation? If a community health worker program increases vaccination rates by 3 percentage points, does that justify the investment? Your MDES should reflect what matters for policy and practice, not just what’s statistically detectable.
A common mistake is powering your trial for an optimistic effect size—the improvement you hope to see—rather than the minimum effect that would be practically important. Be conservative. If your effect size estimate could plausibly range from 10% to 30%, power for something closer to 10%.
Figure 10.1 illustrates this tradeoff using a concrete example. Suppose you’re designing a trial of psilocybin therapy for treatment-resistant depression, with the Montgomery-Åsberg Depression Rating Scale (MADRS, scored 0–60) as your primary outcome. Before the trial, you decide that a 3-point difference on the MADRS is the smallest improvement that would be clinically meaningful—enough to justify the cost and complexity of treatment. As your sample size grows, the MDES shrinks—but it never reaches zero. The shaded band shows a realistic recruitment range, and the dashed line marks the 3-point threshold. The question isn’t “can I reach statistical significance?” It’s “can I detect an effect worth acting on, given the sample I can realistically recruit?”

You can also flip the question: instead of “how small an effect can I detect at this sample size?”, ask “how large a sample do I need to detect the effect I care about?” Figure 10.2 shows this inverted view for the same MADRS example. It makes the starting point explicit—begin with the effect that would change practice, then read off the sample size you need.

In practice, sample size calculations also require assumptions about outcome variability, the chosen significance level, and the chosen statistical power. For cluster-randomized trials, you’ll need to account for intracluster correlation, which can inflate your required sample size dramatically. And you’ll need to build in buffers for attrition and non-compliance—real-world complications that every global health trial faces.
For complex designs—cluster trials, factorial designs, adaptive trials—closed-form sample size formulas may not exist or may not capture the design’s nuances. In these cases, simulation-based power analysis is often the better approach: you simulate your trial thousands of times under different assumptions and estimate the probability of detecting your target effect. This is more flexible and often more realistic than plugging numbers into a formula.
Talk to a statistician during study design, not after data collection. Too many global health trials turn out to be hopelessly underpowered because researchers were overly optimistic about effect sizes or didn’t adequately account for the realities of their design.
THE ART AND SCIENCE OF RANDOMIZATION
Now we get to the heart of RCT design: how do you actually randomize participants to intervention and control groups?
Simple randomization is like flipping a coin for each participant. This works fine for large trials—over thousands of participants, you’ll end up with roughly equal groups that are balanced on all characteristics, measured and unmeasured. But in smaller trials, simple randomization can produce imbalanced groups just by chance.
That’s why most global health RCTs use more sophisticated approaches:
Block randomization ensures that you get equal group sizes. You randomize participants in “blocks”—for example, in blocks of four, you might randomly determine which two of each block go to intervention and which two go to control. This guarantees that at the end of the trial, your groups will be exactly equal in size (or very close if your final block isn’t complete). Block randomization is especially useful when enrollment is rolling—you’re recruiting participants over weeks or months and don’t know the final sample size in advance. Blocks guarantee that if the trial stops at any point, the groups are roughly balanced. If you know the entire pool of participants upfront, you can simply randomize with fixed allocation and don’t need blocks.
Stratified randomization ensures balance on key characteristics you know beforehand. Suppose you’re testing a tuberculosis intervention in Peru and you know that TB rates vary substantially across healthcare centers in your study area (Shah et al., 2015). You might stratify randomization by baseline TB case rate, ensuring that both high-burden and low-burden centers are represented equally in intervention and control groups.
Cluster randomization is when you randomize groups rather than individuals—entire villages, schools, or health facilities. This is often necessary when interventions are delivered at the community level or when contamination between individuals would undermine the design. Cluster randomization introduces important complications for sample size and analysis, which we cover in detail in the cluster randomization section later in this chapter.
Figure 10.3 compares these four approaches visually. Each dot is a participant (or, in the cluster panel, a site). Notice how block and stratified randomization impose more structure to guarantee balance—at the cost of added complexity.

BLINDING: WHO KNOWS WHAT, AND WHEN DOES IT MATTER?
Blinding (also called masking) refers to keeping study participants, providers, and/or outcome assessors unaware of who is receiving the intervention versus control condition. The purpose is to prevent knowledge of group assignment from influencing behavior or outcome measurement.
In a single-blind trial, participants don’t know which group they’re in, but providers and researchers do. In a double-blind trial, neither participants nor providers know. In a triple-blind trial, outcome assessors also don’t know.
Blinding isn’t always possible in global health RCTs. Consider a community health worker program—you can’t blind participants to whether they’re receiving home visits from a health worker or not. The intervention is inherently visible. Does that make the trial invalid? No. But it does mean you need to think carefully about how knowledge of group assignment might influence outcomes.
For example, if your outcome is a behavior that participants self-report (like whether they treated their drinking water), lack of blinding could introduce bias. Participants who know they’re receiving the intervention might report more positive behaviors to please researchers, regardless of what they actually do. To address this, you might use objective measures (microbiological testing of water quality) rather than self-reported measures.
In contrast, the Ethiopian cholera vaccine trial used double-blinding, with participants receiving either the vaccine or an identical-appearing placebo (Desai et al., 2015). Neither participants nor the clinical staff administering injections and collecting blood samples knew who received which. This eliminated the possibility that expectations about the vaccine could influence immune response measurement (which was objective anyway) or participant behavior.
Here’s my practical advice: blind when you can, especially for subjective outcomes or when there’s potential for behavior change based on group assignment. But don’t abandon an important RCT just because blinding isn’t feasible. Instead, use objective outcome measures when possible, and carefully assess and report potential biases that could result from lack of blinding.
Table 10.1 summarizes who is kept unaware of group assignment under each blinding scheme. In global health, full double- or triple-blinding is often impossible—but you can almost always blind outcome assessors, and that alone goes a long way. One form of blinding that’s often overlooked is analyst blinding: the statistician analyzing the data doesn’t know which group is “treatment” and which is “control” until the analysis is complete. This prevents conscious or unconscious analytical choices from being influenced by knowledge of which group is which—a particularly valuable safeguard given the many decisions analysts make about model specification, outlier handling, and subgroup analysis.
CHOOSING YOUR CONTROL CONDITION: WHAT’S THE RIGHT COMPARISON?
This might seem obvious—the control group doesn’t get the intervention, right? But it’s actually one of the most consequential decisions you’ll make, because your choice of comparator defines the question your trial answers. An intervention that looks clearly superior against a weaker comparator might be indistinguishable from a stronger one. The estimated effect is always relative to the comparison, not absolute.
The FDA recognizes five types of controls as valid for “adequate and well-controlled” trials (21 CFR 314.126), and the same logic applies to global health research:
Placebo concurrent control: Participants receive an inert substance or sham procedure designed to resemble the intervention as closely as possible. This controls for placebo effects and provides a clean estimate of absolute efficacy. The Ethiopian cholera vaccine trial used this approach—control participants received saline injections (Desai et al., 2015). Placebo controls are appropriate when no proven treatment exists or when withholding treatment is ethical for the condition studied.
No treatment concurrent control: The intervention group is compared with a group that receives no treatment at all. This differs from placebo control in that participants know whether they are receiving treatment. It’s appropriate when objective outcome measures are available and the placebo effect is negligible—for example, when measuring all-cause mortality or laboratory values that aren’t influenced by participant expectations.
Active treatment concurrent control: Participants receive a known effective therapy rather than placebo or no treatment. This is appropriate when withholding all treatment would be unethical because an effective intervention already exists. The low-osmolarity ORS trials compared a new formulation against standard ORS, not against no treatment (Zubairi et al., 2024). A key challenge: if the new treatment and the active control produce similar results, you can’t always tell whether both worked or neither did.
When an active comparator is used, a critical question is whether it should be any licensed treatment or the best available treatment. Superiority over a weaker comparator is a real finding, but it may not answer the question that matters to patients and policymakers. In global health, “standard of care” varies dramatically by setting, making this choice especially consequential.
Dose-comparison concurrent control: At least two doses of the same intervention are compared, sometimes alongside a placebo or active control. This is useful when the dose-response relationship itself is the question—common in early-phase drug trials. If the drug is effective, different doses should produce different magnitudes of response.
Historical control: Results from the intervention group are compared with prior experience—either the documented natural history of the disease or outcomes from previously treated patients. The FDA considers this the weakest of the five designs because it’s difficult to ensure that the treated group and the historical reference group are truly comparable. Historical controls are generally reserved for diseases with high and predictable mortality or situations where the drug effect is self-evident.
Beyond these five FDA-recognized types, two additional control designs appear frequently in global health research:
Standard care control: Participants receive whatever care they would normally receive in routine practice. This is common in effectiveness trials because you’re testing whether your intervention improves outcomes compared to the status quo. The tuberculous meningitis trial in Vietnam used this approach in a double-blind, placebo-controlled design: participants received adjunctive aspirin (at low or high dose) or matched placebo, all on top of standard anti-tuberculosis drugs and dexamethasone (Mai et al., 2018).
Waiting-list control: Participants are randomized to receive the intervention either immediately or after a delay. This addresses the ethical concern of completely withholding a potentially beneficial intervention, while still allowing comparison between groups. Many behavioral intervention trials use this design.
The choice among these options depends on your research question, ethical considerations, and what you need to demonstrate. If you’re trying to prove that your intervention has any effect beyond placebo, use placebo control. If you’re trying to prove that your intervention improves on current practice, use standard care or active control. If you’re evaluating dose-response, use dose-comparison.
There’s no single right answer, but there is a right process: think explicitly about what comparison will answer the most important question for policy and practice in your setting. The next time you’re reading a trial—or designing one—stop and ask: what is this being compared against, and why? The answer shapes everything that follows.
MEASUREMENT AND ENDPOINTS: WHAT WILL YOU MEASURE?
Before you finalize your trial design, you need to decide what you’re going to measure—and this decision shapes everything from sample size to analysis plan to policy relevance. We cover measurement in depth in Chapter 9, so here I’ll focus on the core decisions specific to RCTs.
Every RCT needs a clearly defined primary endpoint—the single outcome that will determine whether the intervention is deemed successful. This is the outcome you power your study to detect, the one you preregister, and the one that drives your main conclusion. Choose it carefully: it should be clinically or practically meaningful, measurable within your study’s timeframe, and responsive enough that your intervention could plausibly change it.
You should also specify secondary endpoints that provide mechanistic insight or capture additional benefits and harms. The CHoBI7 mobile health trial in Bangladesh chose diarrhea prevalence in children under five as its primary endpoint, but also measured child growth, WASH practices, and water quality—outcomes that help explain how the intervention worked (George et al., 2021).
Caution on multiple endpoints: Every additional outcome you test increases the probability of finding a statistically significant result just by chance (the multiple comparisons problem). Specify in advance which endpoint is primary and which are secondary, and adjust your statistical analyses accordingly.
A few practical considerations:
Objective vs. self-reported measures. In global health settings, you’ll often need to balance the desire for objective, gold-standard measurement against practical constraints. Microbiological confirmation of tuberculosis is more accurate than symptom-based diagnosis, but if your study site doesn’t have laboratory capacity, you may need clinical endpoints instead. Directly observed behaviors are better than self-reports, but observation at scale may not be feasible. Be transparent about these tradeoffs.
Surrogate vs. clinical endpoints. Sometimes you’ll measure a biomarker (antibody titers, viral load) rather than a clinical event (disease, death). Surrogate endpoints can shorten trial duration and reduce sample size requirements, but they only matter if they reliably predict the clinical outcome you actually care about.
Timing matters. When you measure your endpoint can be as important as what you measure. An intervention might show effects at 3 months that fade by 12 months—or vice versa. Think carefully about the biological or behavioral timeline of your intervention and measure at time points that capture meaningful change.
10.3 Trial Registration
Before you enroll your first participant, you should register your trial. This is essential for scientific integrity, and increasingly it’s a regulatory requirement.
WHAT COUNTS AS A “CLINICAL TRIAL”?
Not every study involving human participants is a clinical trial. The NIH defines a clinical trial as a research study in which one or more human subjects are prospectively assigned to one or more interventions to evaluate the effects of those interventions on health-related biomedical or behavioral outcomes. The key elements are prospective assignment to an intervention and a health-related outcome.
The NIH narrowed its interpretation in 2026, clarifying that basic experimental studies with humans (BESH)—like brain imaging or cognitive psychology experiments—are not clinical trials, even though they involve human participants and experimental manipulation. The distinction rests on intent: is the study trying to evaluate whether an intervention affects health, or to understand fundamental biological or behavioral processes?
This matters because the definition determines your regulatory obligations. If your study meets the clinical trial definition, you must register it, comply with reporting requirements, and in many cases follow additional regulatory oversight. The WHO, ICMJE, and EU each draw the line slightly differently, but all require prospective assignment to an intervention evaluated against a health outcome. If you’re running an RCT of any kind in global health, your study almost certainly qualifies.
WHY REGISTER?
Trial registration requires you to publicly commit to your primary outcomes, sample size, and analysis plan before you see your data. This prevents several problems that have plagued the biomedical literature:
Outcome switching: Without pre-registration, researchers might collect data on 20 different outcomes, find that only one shows a significant effect, and then report that one as if it were the primary outcome they’d always planned to study.
Selective reporting: Trials with negative results are less likely to be published than trials with positive results. Registration creates a public record of all trials conducted, making selective non-publication harder to hide.
P-hacking: Without pre-commitment to an analysis plan, researchers might try multiple analytical approaches and report whichever one yields p < 0.05.
Going beyond registration, consider pre-registering a detailed pre-analysis plan (PAP). This document specifies exactly how you’ll handle missing data, what subgroup analyses you’ll conduct, how you’ll adjust for multiple comparisons, and what sensitivity analyses you’ll run. Pre-registration doesn’t lock you into only those analyses—you can still conduct exploratory analyses. It just requires you to transparently distinguish between pre-specified confirmatory analyses and post-hoc exploration.
WHERE TO REGISTER
ClinicalTrials.gov is the most widely used registry and the one required by U.S. federal law for trials subject to FDA regulation. It’s also accepted by the ICMJE for publication purposes. Other WHO primary registries—including the EU Clinical Trials Register, ISRCTN, and regional registries—also meet journal requirements.
The Declaration of Helsinki (2013, Paragraph 35) calls for prospective registration of all research involving human subjects—not just drug trials. This ethical benchmark is broader than most regulatory requirements.
For trials that don’t fall under regulatory mandates but where you still want a public record of your study design—a good practice regardless—platforms like OSF Registries accept preregistrations for any study type, and the AEA RCT Registry covers randomized controlled trials in economics and development research. The key is having a time-stamped, public record of what you planned before you saw the results.
10.4 Ethical Considerations in Global Health RCTs
RCTs raise ethical questions that go beyond standard research ethics—questions we’ll explore more fully in a later chapter on research ethics. Here I want to focus on three issues that are specific to randomized trials: the ethics of withholding treatment from a control group, why randomization is often more ethical than the alternatives in resource-constrained settings, and the ethics of testing interventions on people who may never benefit from them.
EQUIPOISE AND THE ETHICS OF WITHHOLDING TREATMENT
Is it ethical to randomly assign people to a control group that won’t receive a potentially beneficial intervention?
This question becomes particularly acute when we’re testing interventions in settings where standard care is minimal or non-existent. If you’re conducting an RCT of a new drug for tuberculosis meningitis in Vietnam, participants in the control group still receive the standard TB treatment plus corticosteroids (Mai et al., 2018). But if you’re testing a community health worker program in a region with no existing health services, participants randomized to the control group may receive nothing.
International research ethics guidelines address this through the principle of equipoise: It’s ethical to randomize people between treatment options only when there is genuine uncertainty in the scientific community about which option is better. If we already know that an intervention works, it’s unethical to withhold it for the sake of research. But if we genuinely don’t know whether the intervention will help—if expert opinions differ, or if prior evidence is mixed, or if we’re testing a new approach without prior evidence—then randomization is ethically justified because we’re not depriving anyone of a known benefit.
But equipoise gets complicated in global health. There might be uncertainty about whether an intervention works in this particular setting, even if it’s proven to work elsewhere. The oral cholera vaccine trials in Haiti and Ethiopia illustrate this (Desai et al., 2015; K et al., 2016). The vaccine had already been shown to work in Asia, but there was genuine uncertainty about effectiveness in Haiti (a non-endemic setting experiencing an outbreak) and immunogenicity in Ethiopia (a different population with different baseline immunity). So randomization was justified by setting-specific equipoise, even though the intervention’s efficacy in other contexts was established.
Several design strategies can mitigate this tension. Standard-of-care control ensures everyone receives at minimum whatever care they would receive outside the study. Waiting-list control designs ensure everyone eventually receives the intervention. And as we discussed earlier, the choice of comparator matters: the goal is that research participation doesn’t leave anyone worse off than they would be otherwise.
OVERSUBSCRIPTION: WHY RANDOMIZATION CAN BE THE FAIREST OPTION
There’s a common objection to RCTs in development and humanitarian settings: “How can you randomly deny people access to a program that might help them?” But this objection often misses a crucial reality. In most of these settings, demand for services vastly exceeds supply. Programs have fixed budgets, limited staff, and capped enrollment. Not everyone who could benefit will be served. Someone is often left out.
The question isn’t whether some people will go unserved. In many cases, they will. The question is how you decide who gets access and who doesn’t.
Without randomization, that decision is usually made through channels that are far less fair: political connections, geographic convenience, first-come-first-served enrollment that favors those with transportation and information, or staff discretion that may reflect unconscious biases. These allocation methods are rarely transparent and almost never evaluated.
Oversubscription—the situation where more people are eligible for a program than it can serve—creates a natural and ethical opportunity for randomization. When an agency can only enroll 500 families out of 2,000 who qualify, a lottery is arguably the fairest allocation mechanism: every eligible family has an equal chance of being selected, the process is transparent, and no one is denied access based on who they know or where they happen to live. The families not selected through the lottery aren’t worse off than they would have been without the study. They would have been excluded anyway, just through a less equitable process.
The International Rescue Committee’s Airbel Impact Lab has built one of the strongest track records of rigorous RCTs in humanitarian settings, including studies of education programs in refugee camps, nutrition interventions in conflict zones, and cash transfer programs in crisis-affected communities. Oversubscription is central to their approach: when programs can’t reach everyone, randomization provides both a fair allocation mechanism and an opportunity to learn what works.
This logic extends beyond social programs to health interventions. A clinic distributing a limited supply of insecticide-treated bed nets, a government rolling out a new vaccine in phases, a humanitarian organization offering mental health services in a refugee camp. All face the same basic constraint: more need than capacity. In each case, randomization converts an unavoidable allocation decision into an opportunity to generate rigorous evidence about whether the intervention actually works, while treating potential beneficiaries more fairly than the status quo.
This doesn’t make every oversubscription situation appropriate for an RCT. You still need equipoise, ethical review, informed consent, and a genuine research question. But it does dismantle the assumption that randomization in resource-limited settings is inherently exploitative. Often, it’s the opposite: it’s the most transparent and equitable way to distribute scarce resources while learning whether those resources are being used effectively.
TESTING ON PEOPLE WHO WON’T BENEFIT
A different ethical problem arises when the populations enrolled in trials won’t have access to the interventions being tested, even if those interventions prove effective.
In the 1990s, researchers conducted HIV prevention trials in sub-Saharan Africa testing short-course AZT regimens to prevent mother-to-child transmission. Some trials used placebo controls even though a longer AZT regimen was already standard of care in wealthy countries. The rationale was that in these African settings, no treatment was the local standard of care. The trials generated valuable evidence, but critics argued that researchers were using a double standard, conducting trials in Africa that would never have been permitted in Western countries.
The Declaration of Helsinki (2013, Paragraph 34) states that participants who benefit from an intervention in a trial should have post-trial access to it. But enforcement is inconsistent, and “access” can mean very different things in practice.
This controversy prompted a deeper question that persists today: if a trial demonstrates that an expensive intervention works, but the population where it was tested can’t afford it, who benefited from the research? The answer is often: patients in wealthier countries whose regulatory agencies needed the evidence for approval.
This isn’t an argument against conducting trials in low-resource settings. It’s an argument for designing trials that answer questions relevant to those settings, testing interventions that could actually be implemented there, and planning for post-trial access from the start. When you’re designing an RCT, ask yourself: if this intervention works, will the people who participated in the trial—or people like them—actually benefit?
10.5 Implementing and Monitoring an RCT
You’ve designed your study, navigated the ethical complexities, and received all necessary approvals. Now you actually have to do the study. That means enrolling participants, delivering interventions, collecting data, and monitoring quality. This section focuses on the practical realities of RCT implementation in global health settings.
IMPLEMENTATION FIDELITY: DID YOUR INTERVENTION ACTUALLY HAPPEN?
Sometimes RCTs fail not because the intervention doesn’t work, but because the intervention was never actually delivered as intended.
This is the challenge of implementation fidelity—ensuring that the intervention is delivered consistently and as designed to all participants randomized to receive it. In pharmaceutical RCTs, fidelity is relatively straightforward: You can verify that participants took their pills by measuring blood levels of the drug. But in complex behavioral interventions or health system changes, fidelity is much harder to ensure and verify.
The CHoBI7 trial in Bangladesh provides a good example of thinking seriously about implementation (George et al., 2021). The intervention involved delivering health education through multiple channels: facility-based counseling, mHealth messages, and home visits. The research team tracked implementation outcomes including:
Reach: What proportion of enrolled households actually received each intervention component?
Dose: How many mHealth messages were delivered? How many home visits occurred?
Fidelity: Were intervention messages delivered as intended, covering all key topics?
Acceptability: Did participants find the intervention acceptable and useful?
By systematically measuring these implementation outcomes, the researchers could distinguish between “the intervention doesn’t work” (participants received it as intended but outcomes didn’t improve) and “the intervention wasn’t delivered properly” (participants didn’t actually receive the full intervention, so of course outcomes didn’t improve).
This kind of process evaluation—systematic assessment of intervention delivery and mechanisms—should be embedded in every global health RCT. Without it, you’re left with profound uncertainty when interpreting negative results. Did the intervention fail because the underlying theory was wrong? Because the intervention was poorly designed? Or because implementation challenges prevented the intervention from ever really happening?
WHEN THINGS DON’T GO ACCORDING TO PLAN
No matter how carefully you design your RCT, unexpected challenges will arise. Equipment breaks. Staff leave. Enrollment proceeds more slowly than anticipated. Communities have concerns you didn’t anticipate. Stuff happens.
Some challenges require formal protocol amendments, changes to your study design that must be reviewed and approved by ethics committees. If enrollment is much slower than expected, you might need to extend the study duration, add study sites, or revise your inclusion criteria. Other challenges require adaptations that don’t change the core design but improve feasibility. In either case, the key principles are transparency and documentation: what challenges arose, how you responded, and what implications your adaptations might have for interpreting results.
Something that every experienced global health researcher knows but rarely gets said in methods textbooks: sometimes the most ethical decision is to stop a study that’s not working. If you realize that the intervention can’t be delivered as designed, or that participant retention is so poor that your results will be hopelessly biased, it may be better to acknowledge failure, document lessons learned, and reallocate resources rather than completing a flawed trial just because you’ve already invested so much.
MONITORING, QUALITY ASSURANCE, AND THE DSMB
If you wait until data collection is complete to examine your data quality, you’ve waited too long. Effective RCTs build in continuous monitoring to detect and address problems while the study is ongoing. But there’s an important distinction between monitoring process and monitoring outcomes. Getting this wrong can compromise both your blinding and your statistical validity.
Regular data quality checks: Review completed data forms or electronic data weekly (at minimum) to identify missing data, implausible values, or patterns suggesting data fabrication. These checks should focus on data completeness, adherence to protocols, and operational quality—not on outcome data by treatment group. The study team can and should monitor enrollment rates, follow-up completion, and data quality without breaking the blind.
Site monitoring visits: If your study involves multiple sites, regularly visit each site to observe data collection procedures, review source documents, and provide feedback to site staff.
Monitoring of adverse events: Track and report any serious adverse events (deaths, hospitalizations, serious health problems) that occur among study participants, regardless of whether they seem related to the intervention.
Looking at outcome data by treatment arm is a different matter entirely. In a frequentist framework, every look at the outcome data increases the probability of a false positive. This is why examining outcome data is the province of the Data Safety Monitoring Board (DSMB), an independent committee typically composed of clinicians, biostatisticians, and ethicists who are not involved in the trial. The DSMB sees unblinded outcome data that the investigators do not, and reviews it at pre-specified interim analyses using statistical methods (like O’Brien-Fleming boundaries) that account for the repeated testing. The DSMB has the authority to recommend stopping the trial early for three reasons: clear benefit has been demonstrated and continuing would be unethical (as happened in the male circumcision HIV prevention trials), unexpected harm has emerged, or futility analyses suggest no benefit will be found even with full enrollment.
A DSMB protects participants and the integrity of the science. Its members see unblinded data that the investigators don’t, and they bear the responsibility of deciding whether the trial should continue. This independence is critical: investigators have strong incentives to continue their own trials, which is exactly why the stopping decision shouldn’t be theirs alone.
This raises a statistical subtlety. Every time you look at outcome data and potentially stop the trial, you increase the chance of a false positive. If you test your data five times during a trial at \(\alpha\) = 0.05 each time, your overall false positive rate climbs to about 14%, not 5%. Group sequential methods solve this by adjusting the significance threshold at each interim look. The most common approach, O’Brien-Fleming boundaries, uses very stringent criteria early (when data are sparse, you might need p < 0.0001 to stop) and more relaxed criteria later (the final analysis uses something close to p < 0.05). This way, the overall Type I error rate stays at 5% across all the looks combined. Figure 10.4 shows how these boundaries work in practice.
Group sequential methods are a frequentist approach—they spend a fixed alpha budget across pre-planned looks. A Bayesian alternative updates the probability that the treatment works at each interim and stops when that probability crosses a threshold (e.g., >99% chance of benefit, or <5% chance of meaningful effect). Bayesian monitoring is more flexible and arguably more intuitive, but it requires specifying prior beliefs.

The male circumcision trials in Africa provide one of the most striking examples of ethical stopping in global health. In the early 2000s, three independent RCTs tested whether voluntary medical male circumcision reduced HIV acquisition in men: one in Orange Farm, South Africa (Auvert et al., 2005), one in Kisumu, Kenya (Bailey et al., 2007), and one in Rakai, Uganda (R. H. Gray et al., 2007). The South African trial was stopped first. At a planned interim analysis in 2005, the DSMB found a 60% reduction in HIV incidence among circumcised men—an effect so large that continuing to randomize men to the control group would mean knowingly withholding a protective intervention. The Kenya and Uganda trials followed shortly after, stopped by their own DSMBs after showing 53% and 51% reductions respectively.
Three independent trials, three independent DSMBs, three decisions to stop early—all converging on the same conclusion. This convergence led WHO and UNAIDS to recommend VMMC as an HIV prevention strategy in 2007 (WHO/UNAIDS, 2007).
Stopping can also go the other direction. The HVTN 702 HIV vaccine trial in South Africa was stopped for futility when the independent data monitoring committee found no evidence of efficacy (G. E. Gray et al., 2021). In both cases—stopping for benefit and stopping for futility—the DSMBs faced a genuine tension between gathering more data and acting on what they already knew. The statistical machinery of group sequential methods exists precisely to make these decisions rigorous rather than arbitrary.
Setting up a DSMB requires thought. You need to recruit independent experts willing to serve and establish a charter specifying when interim analyses will occur and what criteria will trigger stopping recommendations. For smaller trials or those with lower-risk interventions, a simpler safety monitoring plan may suffice. But for any trial where serious adverse events are plausible, independent safety monitoring is not optional.
REPORTING YOUR TRIAL: THE CONSORT STATEMENT
When it’s time to write up your results, you need to report them in a way that lets readers evaluate the quality and validity of your findings. The CONSORT (Consolidated Standards of Reporting Trials) statement provides an evidence-based minimum set of recommendations for reporting RCTs.
CONSORT exists because systematic reviews have shown that poor reporting makes it difficult to assess trial validity and replicate findings. A trial might be well-designed and carefully conducted, but if the published paper doesn’t describe the randomization process, doesn’t report a flow diagram of participant enrollment, or doesn’t present baseline characteristics of the groups, readers can’t tell whether the results are trustworthy.
CONSORT has two core components: the flow diagram and the checklist.
The flow diagram tracks participant movement through the trial—from initial assessment for eligibility, through randomization, allocation, follow-up, and final analysis. This single figure tells readers more about the trial’s integrity than pages of prose. How many people were screened? How many were excluded, and why? How many were lost to follow-up in each arm? How many were included in the final analysis? When these numbers don’t add up, you know something went wrong. Figure 10.5 shows the flow diagram from the landmark ANRS 1265 trial, which randomized 3,274 uncircumcised men in Orange Farm, South Africa to evaluate whether male circumcision reduced HIV acquisition risk (Auvert et al., 2005).
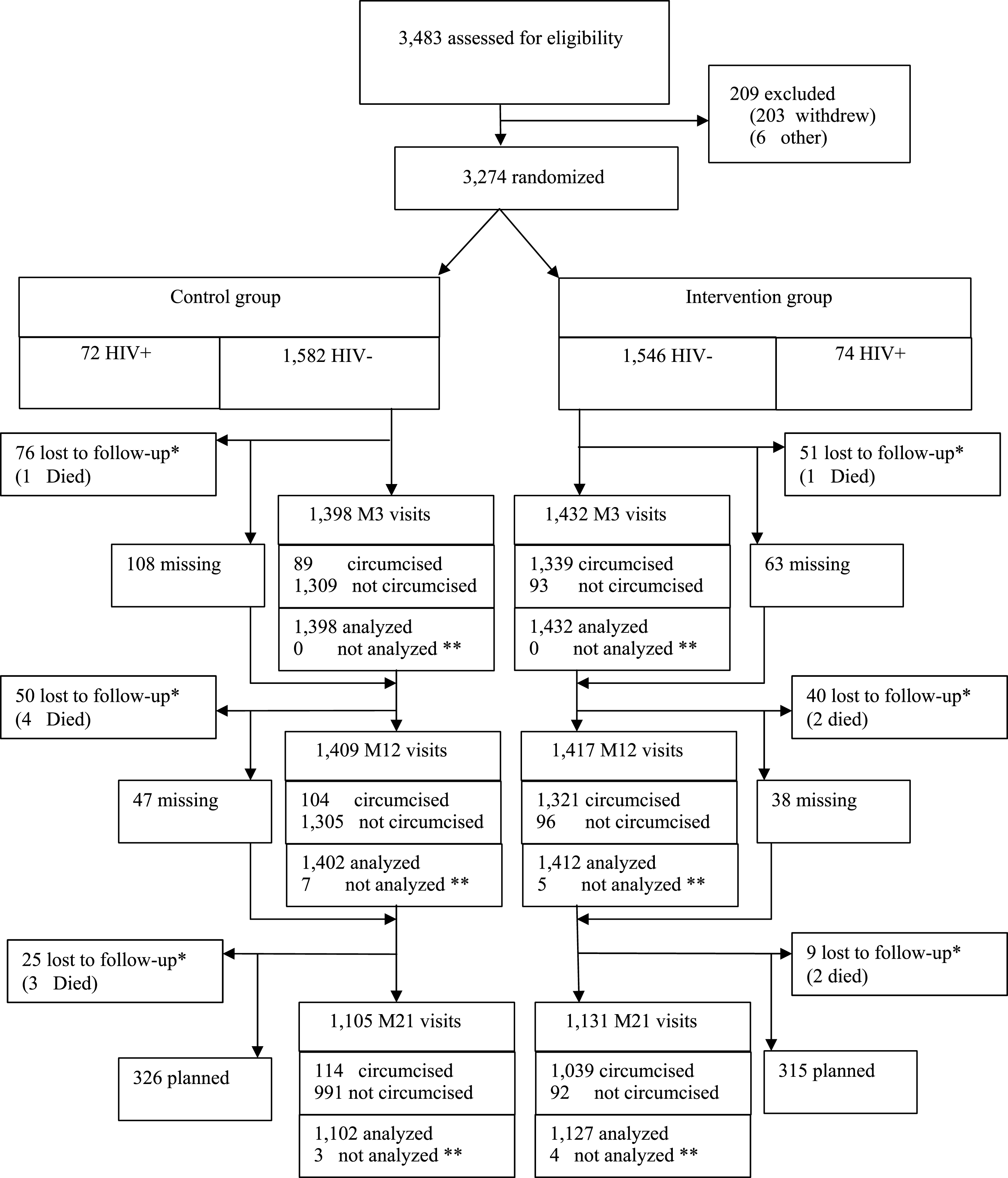
The CONSORT checklist is a 25-item list covering everything a reader needs to evaluate your trial. Many journals require it as a supplementary appendix at submission. The items span the full arc of a trial report:
- Title and abstract: Did you identify the study as a randomized trial? (You’d be surprised how often this is missing.)
- Introduction: Scientific background, rationale, and specific objectives or hypotheses.
- Methods: Detailed description of the trial design, participants, interventions, outcomes, sample size determination, randomization (sequence generation, allocation concealment, implementation), and blinding.
- Results: Participant flow (the diagram), baseline characteristics of each group, numbers analyzed, primary and secondary outcomes with effect estimates and confidence intervals, and harms.
- Discussion: Interpretation of results, limitations, generalizability, and registration information.
The checklist is available at consort-statement.org. I’d recommend downloading it before you start writing up your results—it’s much easier to collect the right information prospectively than to reconstruct it after the fact.
CONSORT also has extensions tailored to specific trial types common in global health:
- Cluster randomized trials—additional items on how clustering was handled in design and analysis
- Non-inferiority and equivalence trials—reporting of margins and justification
- Pragmatic trials—reporting on the PRECIS-2 framework dimensions
- Social and psychological interventions—adaptations for behavioral research
- CONSORT-Equity—reporting standards for health equity trials, including how equity-relevant factors were considered in design, analysis, and interpretation
10.6 Threats to Validity in Practice
Randomization gives you a fair comparison at baseline. But between randomization and analysis, things go wrong. Participants drop out. Others don’t take the treatment they were assigned. And the very act of being studied can change behavior. These threats don’t invalidate the RCT, but ignoring them can.
ATTRITION: WHEN PARTICIPANTS DISAPPEAR
Some loss to follow-up is inevitable in any trial. Participants move, lose interest, get sick, or die. The immediate cost is statistical: fewer participants means less power to detect an effect. If you planned for 200 per arm and 30% drop out, you’re now running an underpowered study.
But the real danger isn’t the lost power. It’s bias when attrition is systematic. If sicker participants in the intervention arm drop out because the treatment has side effects, while healthier participants stay, your remaining sample no longer represents the population you randomized. The treatment group looks artificially healthy. The comparison is no longer fair.
The best defense against attrition bias is prevention: convenient scheduling, transportation support, strong relationships with participants, and data collection that’s as brief and non-burdensome as possible.
How do you assess whether attrition is a problem? Start by comparing baseline characteristics of completers versus dropouts in each arm. If dropouts look similar to completers on measured characteristics, and if attrition rates are similar across arms, the threat is lower. If dropouts in the treatment arm look systematically different from dropouts in the control arm, you have a problem that no statistical method can fully solve.
When attrition does occur, sensitivity analysis is your main tool. The most conservative approach is to assume the worst: that everyone who dropped out of the treatment arm had bad outcomes, and everyone who dropped out of the control arm had good outcomes. If your results hold under this extreme assumption, they’re robust. More commonly, you’ll use multiple imputation or inverse probability weighting to account for missing data under various assumptions, and report how sensitive your conclusions are to those assumptions.
NON-COMPLIANCE: WHEN PARTICIPANTS DON’T FOLLOW THE PLAN
Non-compliance takes two forms. Participants assigned to the intervention may not take it up. They skip sessions, don’t take the medication, or never show up at all. Participation is voluntary after all. And participants assigned to control may obtain the intervention on their own. Both dilute the contrast between arms.
How you analyze the data in the face of non-compliance depends on the question you want to answer.
Intention-to-treat (ITT) analysis keeps participants in their randomized group regardless of what they actually did. Someone randomized to the intervention who never showed up? Still analyzed as intervention. This preserves the benefits of randomization and answers a policy-relevant question: what happens when you offer this intervention to a population, knowing that real-world uptake will be imperfect? It’s conservative, but ITT is the standard primary analysis for RCTs and should always be reported.
Per-protocol analysis restricts to participants who actually received the intervention as intended (and control participants who didn’t cross over). This answers a different question: what is the effect in people who actually adhere? The problem is that adherers differ systematically from non-adherers. They tend to be healthier, more motivated, and have better social support. So per-protocol analysis can introduce selection bias, even though it seems to answer a more clinically relevant question.
ITT tells you the effect of the policy (offering the intervention). Per-protocol and CACE tell you the effect of the treatment (receiving it). Both are useful, but ITT should always be primary.
Complier average causal effect (CACE) analysis—sometimes called the treatment effect on the treated—uses instrumental variables to estimate the effect of actually receiving the intervention among people who would comply if offered it. Unlike per-protocol analysis, CACE accounts for the fact that compliance is not random. It uses randomization itself as an instrument: being assigned to the treatment arm only affects your outcome through receiving treatment (if you’re a complier). This approach recovers a causal estimate, but only for the subpopulation of compliers, not for everyone.
Most RCTs should report ITT as the primary analysis, with per-protocol and (where relevant) CACE analyses as supplementary. Together, they tell a more complete story than any single approach.
EXPERIMENTER EFFECTS: WHEN BEING STUDIED CHANGES BEHAVIOR
Even with perfect retention and full compliance, the act of being in a trial can alter outcomes. Participants who know they’re being observed may change their behavior: eating better, exercising more, adhering to medications more carefully than they would outside the study. This is sometimes called the Hawthorne effect, and it affects both arms equally, making the intervention look less effective relative to control (since control participants also improve). Research staff can also inadvertently influence outcomes through differential attention, enthusiasm, or subtle cues about which arm they believe is better. Good blinding, standardized protocols, and objective outcome measures help, but in behavioral and community-level trials where blinding is impossible, experimenter effects are a reality you need to acknowledge.
10.7 Advanced Topics and Special Considerations
As you become more experienced with RCT design and implementation, you’ll encounter situations that require going beyond the basic principles we’ve covered so far. This section explores some of the more nuanced and challenging aspects of conducting RCTs in global health settings.
CLUSTER RANDOMIZATION: SPECIAL CHALLENGES AND SOLUTIONS
I mentioned cluster randomization earlier, but it deserves deeper exploration because it’s so common in global health and introduces distinct challenges.
Recall that in cluster randomization, you randomize groups (villages, schools, health facilities) rather than individuals. This is necessary when:
- The intervention is delivered at the group level (like a community health worker program)
- Contamination (also known as spillover) between individuals is likely (like an infectious disease intervention)
- Individual randomization would be logistically impossible or culturally inappropriate
Spillover: when treatment crosses group boundaries
That second point—spillover—deserves unpacking, because it’s one of the most underappreciated complications in trial design. Spillover occurs when the treatment given to one person affects the outcomes of another person who wasn’t treated. From a public health perspective, spillover is often the whole point. You want deworming one child to reduce transmission to nearby children. You want vaccinating enough people to create herd immunity. You want health information shared with one household to spread through a community.
But spillover complicates research because it violates the stable unit treatment value assumption (SUTVA): the assumption that one person’s treatment status doesn’t affect another person’s outcome. If treated individuals in the control group’s village share health information they learned, or if deworming children in the treatment group reduces parasite loads for untreated children nearby, then the control group isn’t really untreated. Your estimated treatment effect shrinks. Not because the intervention doesn’t work, but because the comparison group is also benefiting from it.
Spillover can be biological (deworming reduces environmental parasite loads for everyone nearby), informational (a mother trained in nutrition practices teaches her neighbor), or behavioral (seeing one household adopt a new latrine design prompts others to do the same). The classic example is Miguel and Kremer’s study of school-based deworming in Kenya, which found that untreated children in treated schools—and even children in nearby untreated schools—experienced health gains from reduced transmission (Miguel et al., 2004).
This is precisely why cluster randomization helps. By randomizing entire villages or schools rather than individuals, you create a buffer between treatment and control groups. Spillover still happens within clusters, but it’s less likely to cross between clusters, especially when clusters are geographically separated. You can also design studies to explicitly measure spillover by comparing outcomes in untreated individuals who are near treated clusters versus far from them.
The cost of clustering: intracluster correlation
Cluster randomization solves the spillover problem, but it introduces a statistical one. People within the same cluster tend to be more similar to each other than to people in different clusters. Children in the same school share teachers, meals, and environmental exposures. Patients at the same clinic see the same providers. Households in the same village face similar economic conditions. This similarity is captured by the intracluster correlation coefficient (ICC), and it means that adding more people from the same cluster gives you less new information than adding people from different clusters.
To build intuition, consider the extreme case: imagine every person in a cluster is an identical clone. If you’ve already measured one clone’s response to treatment, measuring a second clone tells you nothing new. You already know exactly what they’ll do. Your effective sample size is 1, no matter how many clones you add. That’s an ICC of 1.0. At the other extreme, if people within clusters are no more similar to each other than to people in other clusters (ICC = 0), clustering doesn’t cost you anything statistically. Reality falls somewhere in between, and even modest ICCs can dramatically inflate the sample size you need.
For a more realistic example, suppose you’re testing a school-based nutrition program and your outcome is child height. Children within the same school share environmental exposures, food environments, and socioeconomic characteristics. They’re more similar than children from different schools. If the ICC is 0.05 (meaning 5% of the variation in height is between schools rather than within schools), and you have an average of 50 children per school, your effective sample size is inflated by a “design effect” of 1 + (50-1) \(\times\) 0.05 = 3.45. In other words, you need 3.45 times as many participants in a cluster trial as you would in an individually randomized trial to achieve the same statistical power.
This is why cluster trials require many more participants than individual trials, and why you need to plan for this during sample size calculation. Ignoring clustering leads to underpowered studies and inflated Type I error rates. Figure 10.6 shows how quickly this adds up—with the same total sample size, a cluster trial can detect much less than an individual trial.

Practical considerations for cluster trials
How many clusters do you need? General guidance suggests at least 12-15 clusters per arm, and preferably many more. Trials with only 4-6 clusters per arm have very limited power and are difficult to analyze appropriately. Don’t rely on these rules of thumb though. Simulate or use a proper calculator.
What if clusters vary in size? They usually do. Villages have different populations, schools have different enrollments, clinics serve different catchment areas. You need to account for this variation in your sample size calculations and analysis.
Should you randomize clusters or individuals within clusters? Sometimes you can do both. For example, you might randomize villages to intervention or control, then within intervention villages, randomize households to receive intensive versus standard engagement. This “individually randomized group treatment” design can be very efficient.
How do you analyze cluster trials? You need statistical methods that account for clustering—mixed effects models, generalized estimating equations, or cluster-level analyses. A biostatistician can help you choose the right approach (and you should definitely consult one if you’re doing a cluster trial).
What about consent? In cluster trials, you often need both cluster-level consent (from community leaders or facility administrators) and individual consent from participants. The ethics get tricky when interventions are delivered at the cluster level but outcomes are measured on individuals.
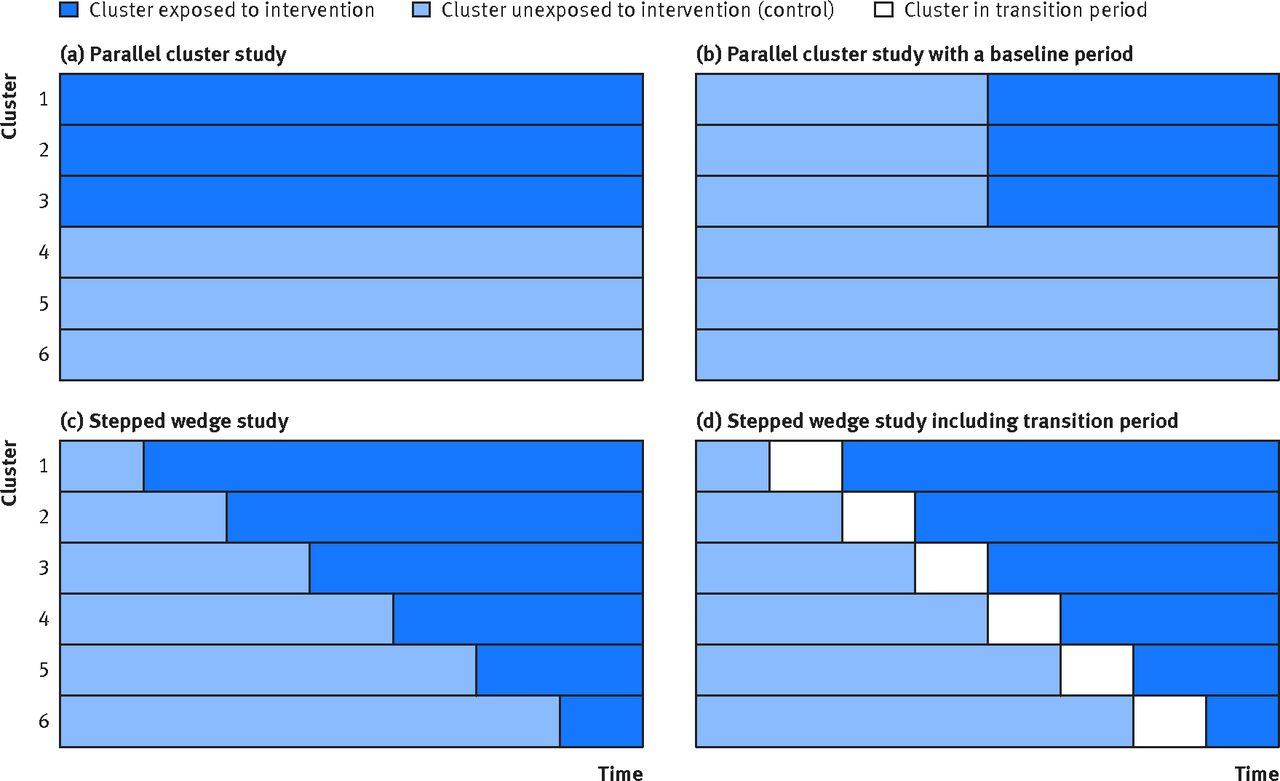
STEPPED-WEDGE DESIGNS: WHEN EVERYONE EVENTUALLY GETS THE INTERVENTION
Standard RCTs maintain distinct intervention and control groups throughout the study. But what if it’s ethically or politically problematic to withhold the intervention from control clusters indefinitely? Or what if you simply can’t turn the intervention on everywhere at once? You have limited trainers, equipment, or funding, and a phased rollout is the only feasible option? If you’re willing to randomize the order in which clusters receive the intervention, a stepped-wedge design becomes a possibility.
In a stepped-wedge design, all clusters start in the control condition and then sequentially cross over to receive the intervention at randomly determined time points. By the end of the study, everyone has received the intervention, but you’ve still been able to make comparisons between intervention and control conditions.
The Peru tuberculosis household contact tracing study I mentioned earlier used this design (Shah et al., 2015). All 34 healthcare centers in the district eventually implemented active case finding for TB contacts, but they started at different randomly determined times over a 20-month period. This allowed the researchers to compare TB diagnosis rates in centers that had already begun active case finding to centers that hadn’t yet started, while respecting the health department’s need to eventually implement the program everywhere.
Stepped-wedge designs have become increasingly popular in global health because they address several practical realities at once. Everyone eventually receives the intervention, which eases ethical concerns about withholding treatment. The phased rollout aligns with how programs actually scale. Few organizations can launch in dozens of sites simultaneously. And communities and policymakers are often more willing to participate when they know they’ll eventually receive the intervention rather than risk being assigned to a permanent control group.
Stepped-wedge designs are vulnerable to time trends—if outcomes shift over time due to seasons, policy changes, or demographics, these temporal changes confound your results. The analysis is also more complex, requiring models for both the stepped rollout and time effects.
Stepped-wedge designs work well when you’re evaluating a program that will be scaled up regardless (so the question is “does it work?” not “should we implement it?”), when sequential rollout is logistically necessary, and when you can plausibly assume that underlying trends are similar across clusters. Figure 10.7 compares the stepped-wedge design to the conventional parallel cluster trial and its variations.
SUPERIORITY, NON-INFERIORITY, AND EQUIVALENCE TRIALS
Most trials you’ll encounter are superiority trials. They test whether an intervention is better than a comparator. But sometimes the goal is to show that a new treatment is not meaningfully worse, perhaps because it’s cheaper, easier to administer, has fewer side effects, or can be manufactured locally. This is called non-inferiority.
Global health is full of non-inferiority questions: testing shorter TB treatment regimens against standard 6-month therapy, comparing community health worker delivery of a service against facility-based delivery, evaluating lower-cost biosimilars against branded biologics, testing heat-stable vaccine formulations against cold-chain-dependent versions. In each case, the question isn’t “is the new approach better?” but “is it good enough that its practical advantages justify adoption?”
The choice of non-inferiority margin is critical and often contentious. It should represent the largest clinically acceptable difference—the point beyond which you’d prefer the original treatment despite any advantages of the new one.
Non-inferiority trials test whether a new treatment’s effect falls within a pre-specified margin of the standard treatment’s effect. If the standard treatment reduces mortality by 30% and you define a non-inferiority margin of 10 percentage points, you’re asking: does the new treatment reduce mortality by at least 20%? If so, you’d consider it “non-inferior”. In other words, close enough in effectiveness that its other advantages (lower cost, simpler delivery) make it worth adopting.
Figure 10.8 illustrates the four possible outcomes using this mortality example. The standard treatment achieves a 30% reduction, and the margin is set at 10 percentage points—so the new treatment must achieve at least a 20% reduction to be considered non-inferior. Everything hinges on where the confidence interval falls relative to two lines: the standard treatment’s effect (30%) and the non-inferiority margin (20%). Only when the entire CI stays to the right of the margin can you claim non-inferiority.

Non-inferiority is a one-sided question: is the new treatment not too much worse? A related but distinct goal is equivalence. Equivalence trials ask a two-sided question: are two treatments close enough in effect that we can consider them interchangeable? Here, you’d reject the null hypothesis if the entire confidence interval falls within the margin on both sides. The new treatment is neither meaningfully better nor meaningfully worse. Equivalence designs are most common in pharmaceutical regulation, where a generic drug manufacturer needs to demonstrate that their product performs the same as the branded version. In global health, you might see equivalence framing when comparing two formulations of the same drug or two diagnostic tests that should yield the same results.
GETTING MORE FROM EVERY TRIAL
Trials are expensive, slow, and logistically demanding—especially in global health settings. A conventional two-arm RCT answers one question: does this intervention work better than the alternative? Given the cost and effort of setting up trial infrastructure, recruiting participants, and following them over time, researchers have developed designs that squeeze more answers out of a single study. These range from testing multiple interventions simultaneously (factorial), to modifying the trial mid-course based on what you’re learning (adaptive), to building perpetual research infrastructure that outlasts any single question (platform trials).
FACTORIAL DESIGNS: TESTING MULTIPLE INTERVENTIONS EFFICIENTLY
Many global health challenges require multi-component interventions. Improving child nutrition might require addressing food security, water quality, disease prevention, and maternal education—not just one of these in isolation. But how do you figure out which components are essential and which are superfluous?
Factorial designs allow you to test multiple interventions simultaneously within a single trial. In a 2x2 factorial design, you randomize participants to four groups: intervention A only, intervention B only, both A and B, or neither. This lets you evaluate the effects of A and B independently, plus whether there’s any interaction between them (whether the combination works better or worse than you’d expect from the individual effects).
The WASH Benefits trials in Bangladesh and Kenya used factorial designs to evaluate water quality, sanitation, handwashing, and nutrition interventions (Luby et al., 2018). Rather than conducting separate RCTs for each component (which would require enormous sample sizes and many years), they tested multiple components and combinations within a single trial. The results were striking: the combined WASH arm significantly reduced diarrhea (PR 0.69), but showed no additive benefit over individual components—sanitation alone (PR 0.61) and handwashing alone (PR 0.60) performed just as well—a finding that challenged the widespread assumption that integrated WASH programs would be more effective than their components and reshaped global WASH policy.
Factorial designs have important advantages: - Efficiency: You can answer multiple research questions within a single trial - Interaction testing: You can determine whether interventions work synergistically or antagonistically - Pragmatism: Many real-world interventions are multi-component, so factorial designs match implementation reality
But they also have limitations: - Complexity: As you add more factors, the design becomes exponentially more complex (a 2x2x2 factorial has eight arms!) - Sample size: You need adequate power for each main effect and potentially for interactions - Interpretation: When interventions interact in unexpected ways, results can be difficult to interpret and communicate to policymakers
My advice: Consider factorial designs when you’re testing interventions that are plausibly independent (their effects don’t depend on each other), when you have adequate sample size, and when you’re prepared for the analytical complexity. But don’t use factorial designs just because they seem efficient—sometimes separate trials testing one thing at a time provide clearer answers.

ADAPTIVE TRIAL DESIGNS
Factorial designs answer more questions, but they’re still locked in from the start. Adaptive trial designs go further by building in pre-planned decision points where the trial can be modified based on accumulating data (Park et al., 2021). The key word is pre-planned. These aren’t ad-hoc changes made because you didn’t like your interim results. They’re adaptations written into the protocol from the start, with statistical methods that preserve the trial’s validity.
These modifications can take several forms: dropping ineffective treatment arms and reallocating new participants to more promising ones, re-estimating the needed sample size based on observed variability, or combining what would traditionally be two separate trial phases into a single seamless design. The 9-valent HPV vaccine trial, for instance, began by testing three dosage levels, selected the most promising dose at an interim analysis, and seamlessly continued into a confirmatory trial, cutting years off the timeline (Joura et al., 2015).
Adaptive designs are particularly valuable in global health when resources are limited, when there’s genuine uncertainty about dosing or intervention delivery, or when multi-arm trials need the flexibility to focus resources on what’s working. But they require more statistical expertise upfront and more robust data systems, so they’re not always the right choice (Park et al., 2021).
PLATFORM AND UMBRELLA TRIALS
Platform and umbrella trials take the logic of “do more with one trial” to its furthest point. These designs fall under the banner of master protocols—a single overarching protocol designed to evaluate multiple interventions or multiple populations (or both) (Park et al., 2021). The key distinctions come down to what varies and what stays fixed.
A platform trial tests multiple interventions for the same condition against a shared control group. The defining feature is that the infrastructure is perpetual: individual treatment arms have their own interim analyses and can end, but the trial itself keeps running, and new arms can be added at any time. This is different from interim stopping in a conventional trial, where the DSMB’s decision ends the entire study. In a platform trial, answering one question is just Tuesday.
The RECOVERY trial is the landmark example. It enrolled over 47,000 hospitalized COVID patients and evaluated dozens of treatments within a single protocol. The hydroxychloroquine arm had its own interim analysis, found no benefit, and was closed—but RECOVERY kept enrolling. The dexamethasone arm found clear mortality benefit, reported it, and changed clinical practice worldwide—but RECOVERY kept enrolling. New treatments were added as they became available, each evaluated against the same shared control group within the same infrastructure.
For more on the RECOVERY trial, see recoverytrial.net.
An umbrella trial also focuses on a single disease, but it stratifies patients into subgroups—usually defined by biomarkers—and tests a different targeted therapy in each subgroup. Everyone has the same disease, but they get different treatments based on their biology. This design is common in oncology, where patients are sorted by tumor mutations and matched to targeted agents.
A basket trial goes the other direction: one targeted therapy is tested across multiple diseases that share a common molecular feature. The therapy is the constant; the diseases vary.
The advantages of master protocols are substantial. Shared control groups mean fewer total participants are needed. All treatments are evaluated under identical conditions, making comparisons more reliable. And the infrastructure persists even as individual research questions are answered. For global health, platform trials are particularly promising for outbreak response (as RECOVERY demonstrated), neglected tropical diseases where multiple treatments need evaluation, and antimicrobial resistance where new agents constantly enter the pipeline. The infrastructure investment is substantial, but once established, these designs answer questions faster and at lower cost than running separate trials one at a time.
10.8 The Limitations: What RCTs Can’t Tell Us
You’ve now seen the full machinery of RCTs: design, ethics, implementation, randomization schemes, adaptive designs, interim analyses. Before we turn to interpreting and translating results, let’s talk about what RCTs can’t do. They’re powerful, but they’re not the answer to every research question.
RCTs answer narrow questions. They tell us whether a specific intervention, delivered in a specific way, to a specific population, in a specific context, improves specific outcomes. They don’t necessarily tell us why an intervention works, for whom it works best, or how to implement it in different settings.
We might conduct an RCT showing that a community health worker program improves child vaccination rates in rural Rwanda. That’s valuable evidence. But the trial doesn’t tell us which components of the program were essential and which were superfluous—unless we design it with multiple arms, as a factorial, or with other advanced features. And it doesn’t explain the mechanisms. Was it because health workers provided information that changed parents’ beliefs about vaccines? Because they reduced transportation barriers? Because they built trust in the health system? All of the above?
To answer deeper questions like these, we need other research methods alongside our RCT: qualitative interviews to understand mechanisms, process evaluations to identify which components were actually implemented as planned, and cost-effectiveness analyses to determine whether the program is sustainable at scale. The best global health RCTs increasingly embed these complementary research methods within the trial design.
RCTs can be artificial. The very rigor that makes RCTs good at establishing causality can make them poor at capturing real-world complexity. In an RCT, we often provide more intensive follow-up, more careful monitoring, and more resources than would be available in routine practice. We exclude people with complicated medical conditions or social situations that might interfere with the study protocol. We measure outcomes at fixed time points that may not align with how health systems actually operate.
This is the tension between efficacy trials (does the intervention work under ideal conditions?) and effectiveness trials (does the intervention work in real-world practice?). Global health has increasingly moved toward pragmatic effectiveness trials that embed research within routine health service delivery, accepting some loss of experimental control in exchange for greater real-world relevance. But it’s a tradeoff, and different research questions require different positions on this continuum.
RCTs can be expensive and time-consuming. Conducting a rigorous RCT typically requires substantial funding, infrastructure, and time. For some urgent global health questions—like how to respond to an Ebola outbreak or implement a new intervention during a humanitarian crisis—we don’t always have the luxury of waiting five years for RCT results. In these situations, we may need to act based on the best available evidence, even if it comes from observational studies or implementation experience rather than RCTs.
RCTs can be unethical or impractical for some research questions. We can’t randomize some exposures (you can’t randomly assign someone to smoke cigarettes for 20 years to study lung cancer risk). We shouldn’t withhold interventions we already know to be effective (it would be unethical to conduct an RCT of oral rehydration therapy versus no treatment for diarrhea today). And for some global health challenges, randomization may be logistically impossible or politically infeasible.
This is why we need a portfolio of research methods. RCTs are invaluable for testing whether interventions work, but we also need observational epidemiology to identify risk factors, qualitative research to understand complex social dynamics, implementation science to figure out how to deliver interventions effectively, and systems thinking to address root causes of health inequity.
RCTs are powerful tools, but they’re tools for specific purposes. The key is knowing when an RCT is the right approach for your research question, and when other methods might serve you better.
10.9 From Results to Policy and Practice
But let’s say an RCT is the right choice for your question. You designed your study thoughtfully, implemented it carefully, collected clean data, and conducted rigorous analyses. You even found that your intervention significantly improved the outcome you care about. Success, right?
Well, maybe. Now comes one of the hardest parts: translating your RCT findings into policy and practice in ways that actually improve population health.
UNDERSTANDING HETEROGENEITY: FOR WHOM DOES THIS WORK?
Most RCTs report average treatment effects: the average difference in outcomes between intervention and control groups across all enrolled participants. But average effects can mask important heterogeneity in who benefits.
The tuberculous meningitis trial in Vietnam illustrates this beautifully (Mai et al., 2018). The primary analysis across all 120 participants didn’t show statistically significant benefit from aspirin. But planned subgroup analyses revealed a significant interaction with diagnostic category. Among the 91 participants with microbiologically confirmed TB meningitis, the intervention showed potential benefit: 34% of placebo recipients experienced new infarcts or death by day 60, compared to 15% in the 81mg aspirin group and 11% in the 1000mg aspirin group.
Does this mean aspirin works for confirmed TB meningitis but not for suspected cases? Maybe. Or maybe the study just wasn’t large enough to detect effects in the unconfirmed cases. This is the challenge with subgroup analyses. They can provide important mechanistic insights, but they also risk false positives when you slice your data many different ways.
The key is to distinguish between pre-specified subgroup analyses (planned before you looked at the data, based on biological or theoretical rationale for why effects might differ across subgroups) and post-hoc exploratory analyses (looking for differences after you’ve seen the results). Both can be valuable, but they require different interpretations. Pre-specified subgroup analyses provide stronger evidence. Post-hoc analyses generate hypotheses that need confirmation in future studies.
For global health RCTs, thinking about heterogeneity is especially important because you’re often testing interventions in diverse populations and contexts. An intervention that works on average might work strongly in some subgroups and not at all in others—and for implementation, you need to know which is which.
This connects directly to external validity, which we explored in depth in Chapter 8. The variables that drive heterogeneity within your trial—the effect modifiers—are the same variables that determine whether your findings will travel to new populations. If maternal BMI modifies the effect in your trial, then differences in BMI distributions between your study population and a target population will shape whether your results apply there. Identifying effect modifiers isn’t just about understanding your own trial, it’s about building the evidence base for transporting results to the populations that need them most.
Figure 10.10 shows what this looks like in practice. The diamond at the top is the overall average effect. Statistically significant and policy-relevant. But the subgroup estimates tell a richer story. The intervention works much better for underweight mothers and when started early in pregnancy, and benefits female infants more than males. If you only reported the overall effect, you’d miss who benefits most and who might not benefit at all.

COST-EFFECTIVENESS: CAN WE AFFORD WHAT WORKS?
Estimating an effect size and looking for potential effect modifiers is a lot of work, but it’s only part of the picture. In global health, not every efficacious intervention will be adopted at scale because resources are limited and choices must be made among competing priorities. Cost-effectiveness analysis helps policymakers make these difficult decisions by comparing the health gains from different interventions relative to their costs.
I won’t dive deep into cost-effectiveness methodology here (that’s a separate chapter), but the key point for RCT design is this: If you want your trial evidence to actually influence policy, embed economic evaluation into your RCT design from the start. That way, in addition to saying an Intervention A increases vaccination rates by 10 percentage points compared to Intervention B (the effect size), you can say that the cost of fully vaccinating kids using Intervention A is 40% less than Intervention B (cost-effectiveness).
Some interventions that work very well clinically may not be cost-effective at scale. Others that show modest clinical benefit may be highly cost-effective if they’re inexpensive and prevent costly downstream morbidity. Without economic data, policymakers can’t make informed decisions about resource allocation.
FROM EVIDENCE TO SCALE: WHY MOST INTERVENTIONS FAIL TO LAUNCH
Even when an RCT demonstrates clear benefit and cost-effectiveness, this is where most interventions fail. Not because the evidence was wrong, but because scaling is hard.
The PRECIS-2 framework helps researchers position their trial along a continuum from highly explanatory (ideal conditions) to highly pragmatic (routine care), using nine domains including eligibility, setting, and follow-up intensity.
We already discussed the tension between efficacy and effectiveness: whether an intervention works under ideal trial conditions versus real-world practice. Making pragmatic design choices helps close this gap: broad eligibility rather than tightly screened participants, routine clinical settings rather than research clinics, usual care as the comparator rather than placebo, and outcomes that matter to patients and providers rather than surrogate markers. Global health increasingly needs more pragmatic trials because we have many interventions that work under ideal conditions but unclear evidence about whether they work when implemented through health systems with limited resources.
But even a pragmatic effectiveness trial doesn’t solve the harder problem: scale-up and dissemination across a large population or health system.
The oral cholera vaccine illustrates this well. Initial efficacy trials in Asia established that OCV could prevent cholera under controlled trial conditions. The Haiti demonstration project tested effectiveness in an outbreak setting with operational delivery at scale (K et al., 2016). This evidence led to WHO endorsement of OCV for both endemic and epidemic use. But translating that endorsement into widespread OCV deployment required addressing vaccine supply chains, financing mechanisms, targeting strategies, and integration with other cholera control measures, challenges that extend far beyond what any single RCT can address.
Or consider the HIV “treatment as prevention” evidence from HPTN 052 (Cohen et al., 2012). The RCT established that early ART dramatically reduces HIV transmission in serodiscordant couples, and the finding reshaped subsequent treatment-as-prevention guidance. But achieving population-level impact required implementing test-and-treat strategies that could identify HIV-positive individuals early, link them to care, support ART adherence, and maintain them in treatment long-term. ART coverage during pregnancy in sub-Saharan Africa increased from 33% in 2010 to 69% in 2019, and mother-to-child transmission rates declined accordingly—but progress has been uneven across the region, reflecting differences in health system capacity rather than differences in the underlying evidence (Astawesegn et al., 2022).
This is why we need implementation science alongside our RCTs. Research that specifically addresses the how of delivering interventions at scale in resource-constrained health systems.
10.10 Closing Reflection
Let me bring us back to where we started: a simple mixture of salt, sugar, and water that has saved more than 50 million lives because researchers in South Asia had the courage and rigor to test whether it actually worked.
That’s the core promise of the RCT. Randomization creates the counterfactual we can never directly observe—what would have happened without the intervention—and that counterfactual is what lets us cleanly say causes rather than correlates with. Everything else in this chapter—blinding, sample size calculations, cluster designs, stepped-wedge rollouts—exists to protect that counterfactual from the messiness of the real world.
But the ORT story also reveals what RCTs can’t do on their own. The trials showed that a salt-sugar-water solution could prevent death from dehydration. They didn’t explain how to train millions of mothers to mix it correctly, or how to sustain supply chains across rural Bangladesh, or how to overcome physician resistance to something that seemed too simple to work. Getting from “the trial showed it works” to “more than 50 million lives saved” required decades of implementation science, community health worker programs, policy advocacy, and political will—work that no trial design can replace.
So hold both truths at once. RCTs are our strongest tool for establishing that an intervention causes the outcomes we care about. And they are only the beginning of the work required to turn that evidence into better health.
Anonymous. (1978). Water with sugar and salt. The Lancet, 2(8084), 300–301.
Astawesegn, F. H. et al. (2022). Trends and effects of antiretroviral therapy coverage during pregnancy on mother-to-child transmission of HIV in sub-saharan africa. Evidence from panel data analysis. BMC Infectious Diseases.
Auvert, B. et al. (2005). Randomized, controlled intervention trial of male circumcision for reduction of HIV infection risk: The ANRS 1265 trial. PLoS Medicine, 2(11), e298.
Bailey, R. C. et al. (2007). Male circumcision for HIV prevention in young men in Kisumu, Kenya: A randomised controlled trial. The Lancet, 369(9562), 643–656.
Cohen, M. S. et al. (2012). HIV treatment as prevention and HPTN 052. Current Opinion in HIV and AIDS.
Coursey, K. et al. (2024). Acceptability of HPV vaccination for cervical cancer prevention amongst emerging adult women in rural mysore, india: A mixed-methods study. BMC Public Health.
Desai, S. N. et al. (2015). A randomized, placebo-controlled trial evaluating safety and immunogenicity of the killed, bivalent, whole-cell oral cholera vaccine in ethiopia. The American Journal of Tropical Medicine and Hygiene.
Fontaine, O. et al. (2007). Oral rehydration therapy: The simple solution for saving lives. BMJ, 334(Suppl 1), s14.
George, C. M. et al. (2021). Effects of a water, sanitation, and hygiene mobile health program on diarrhea and child growth in bangladesh: A cluster-randomized controlled trial of the cholera hospital-based intervention for 7 days (CHoBI7) mobile health program. Clinical Infectious Diseases.
Government of India, Ministry of Health and Family Welfare. (2011). Final report of the committee appointed by the govt. Of india to enquire into alleged irregularities in the conduct of studies using human papilloma virus (HPV) vaccine by PATH in India. Government Inquiry Report.
Gray, G. E. et al. (2021). Vaccine efficacy of ALVAC-HIV and bivalent subtype C gp120-MF59 in adults. New England Journal of Medicine, 384(12), 1089–1100.
Gray, R. H. et al. (2007). Male circumcision for HIV prevention in men in Rakai, Uganda: A randomised trial. The Lancet, 369(9562), 657–666.
Hemming, K. et al. (2015). The stepped wedge cluster randomised trial: Rationale, design, analysis, and reporting. BMJ, 350, h391.
Joura, E. A. et al. (2015). A 9-valent HPV vaccine against infection and intraepithelial neoplasia in women. New England Journal of Medicine, 372(8), 711–723.
K, S. et al. (2016). Effectiveness of oral cholera vaccine in haiti: 37-month follow-up. The American Journal of Tropical Medicine and Hygiene.
Luby, S. P. et al. (2018). Effects of water quality, sanitation, handwashing, and nutritional interventions on diarrhoea and child growth in rural Bangladesh: A cluster randomised controlled trial. The Lancet Global Health, 6(3), e302–e315.
Mahalanabis, D. et al. (1973). Oral fluid therapy of cholera among Bangladesh refugees. Johns Hopkins Medical Journal, 132(4), 197–205.
Mai, N. T. H. et al. (2018). A randomised double blind placebo controlled phase 2 trial of adjunctive aspirin for tuberculous meningitis in HIV-uninfected adults. Elife.
Medical Research Council Streptomycin in Tuberculosis Trials Committee. (1948). Streptomycin treatment of pulmonary tuberculosis. British Medical Journal, 2(4582), 769–782.
Miguel, E. et al. (2004). Worms: Identifying impacts on education and health in the presence of treatment externalities. Econometrica, 72(1), 159–217.
Nalin, D. R. et al. (2018). 50 years of oral rehydration therapy: The solution is still simple. The Lancet, 392(10147), 536–538.
Park, J. J. et al. (2021). Randomised trials at the level of the individual. The Lancet Global Health, 9(5), e691–e700.
Shah, L. et al. (2015). Implementation of a stepped-wedge cluster randomized design in routine public health practice: Design and application for a tuberculosis (TB) household contact study in a high burden area of lima, peru. BMC Public Health.
Snyder, J. D. et al. (1982). The magnitude of the global problem of acute diarrhoeal disease: A review of active surveillance data. Bulletin of the World Health Organization, 60(4), 605–613.
WHO/UNAIDS. (2007). New data on male circumcision and HIV prevention: Policy and programme implications.
Zubairi, M. B. A. et al. (2024). Low-osmolarity oral rehydration solution for childhood diarrhoea: A systematic review and meta-analysis. Journal of Global Health.